 HUN-REN Biological Research Centre, Szeged
HUN-REN Biological Research Centre, Szeged 


Molecular biophysics approach to bio-medically relevant live processes associated with biomembranes
Our unique spectroscopy-based molecular biophysics ap proach yields functionally relevant data on structure and dynamics of our selected membrane-protein systems. Most of the topics of our own and the collaborating partners are of strong bio-medical relevance. We use localised and transient spectroscopy to tune into the spatial location and time window of native molecular events in biomembranes and membrane-associate proteins. Therefore, our data are fully functionally relevant. The combination of experimental molecular data with molecular mechanics and artificial intelligence (AI) is very promising, and will allow us to build atomistic and physical models of the native biomolecules and their mechanism of action. Our main technique is (site-directed) spin-labelling and spin-trapping electron paramagnetic resonance (EPR) spectroscopy (we have the best X-band continuous wave and pulsed EPR spectrometer in Central- and East Europe). We also use fluorescence and Fourier-transform infrared spectroscopy to study native or attached fluorescent groups and molecular vibrations, respectively, in biomolecules. These techniques are combined with reaction-triggering techniques for studying kinetics. Phase transitions are studied with our differential scanning calorimeter. We also have a computer cluster for the theoretical work. Essentially, our approach is functional structure biology, which we use in many collaborations to study free radicals, model and biomembranes, lipid-based drug-delivery formulas, cell cultures, soluble and membrane proteins and food products.
Our open PhD topics are listed in two Doctoral Schools. Foreign students are encouraged to apply for Stipendium Hungarica fellowship to do PhD work on these topics. Our popular science public outreach channel is on youtube.
Membrane-associated proteins of the pH regulation and membrane fusion processes of the organelles of eukaryotic cells
The molecular mechanisms of membrane fusion and pH regulation of the eukaryotic cellular organelles are of high relevance to many normal and pathogenic physiological processes. We focus on 3 classes of key proteins in these processes: the vacuolar proton-ATPase (V-ATPase), the ductin and syntaxin proteins. The former two are our projects, and they are strongly related since part of the V-ATPase belongs to the ductin family. Ductins and syntaxins play important roles in vesicle fusion mechanisms. Some ductin proteins function as gap-junctions and synaptic mediatophores. We have previously determined the membrane position and interaction of one of the gap-junction ductin proteins with lipids. In collaboration with the Momentum Drosophila Autophagy group (G. Juhász, Institute of Genetics) we are studying the binding of syntaxins to membranes and their interaction with V-ATPase. The internal organelles of eukaryotic cells are more acidic than the cytoplasm. The protein complex that is responsible for their acidification is nature's most universal proton pump, the vacuolar proton-ATPase. V-ATPase also takes part in membrane fusion processes by providing controlled local acidification in cells. V-ATPase plays crucial roles in a number diseases, and its tissue-specific inhibition is a key therapeutic objective. Because of its size and complexity, understanding the details of structure-function relationship of this protein complex is one of the major challenges in biophysics and molecular biology today. V-ATPase is a membrane-bound molecular rotary engine (Fig. 1), which converts the chemical energy from ATP hydrolysis to the rotation of the rotor domain via a torque generated between specific subunits. This leads to trans-membrane proton pumping in the interface between the stator and rotor domains. Our studies on V-ATPase aim at subunit-subunit and subunit-lipid interactions, the effect of synthetic inhibitors and divalent cations on function and subunit assembly and the details of the rotary mechanism. We have recently measured the rate of rotation of the rotor in the yeast V-ATPase in native vacuolar membrane vesicles. We were the first to report the rotation rate in a V-ATPase that is not subjected to genetic or chemical modification and it is not fixed to a solid support. We have also discovered that the activity of V-ATPase can be affected with an oscillating trans-membrane electric field. The electric field effects are being used to explore further details of the rotary mechanism. In reconstituted model membrane system we are studying the functional assembly of the c-ring, the ductin part of V-ATPase.

Figure 1. The rotary mechanism of V-ATPase. Each subunit c binds a proton when in contact with lipids. Protons enter and leave the rotor in hydrophilic input and output channels, between subunits c and a.
Artificial intelligence and molecular mechanics approach in the structure biology of membrane proteins
Classical structural biology of membrane proteins faces the challenges and limitations due to the need of isolating, solubilising or crystallising proteins (that are natively hosted by the lipid matrix) in a foreign environment. It is therefore not surprising that out of the more than 100 thousand known protein structures there are only a few hundreds of trans-membrane membrane proteins, whereas roughly 30% of the genes in the human genome code such proteins, and more than 50% of drugs target membrane proteins. In addition, there is always the question: to what extent the obtained structures and features of isolated membrane proteins correlate with those under native, in-membrane conditions? Theoretical approaches have therefore enormous importance. Fig. 2 shows the essence of the fundamental and most challenging problem of structure biology: if we know the sequence of a protein, how can we predict its structure? Since the native fold of a membrane protein assumes a native lipid annular shell, the sequence-to-fold coding, illustrated in Fig. 2, is valid only in the native membranous environment. Therefore, our approach is to measure structural data on the target membrane proteins in their functional, native environment, and combine the so-obtained data with theoretical methods in order to predict the 3-dimensional fold. A good example is our prediction of the basic fold of the conserved four-helix trans-membrane bundle of the cytochrome b561 membrane protein family: even after more than two decades of failed efforts for crystallising any protein from the family, our published structures were the only ones available for about 10 years. In the past we have "manually" modelled the membrane location of cytochrome c, based on our own proximity measurements in reconstituted bilayer. We have identified the backbone structures of the major coat protein of the M13 bacteriophage that are most compatible with structural data measured in membranes. The main objective of this project is to improve existing protein structure prediction techniques with focus on membrane proteins. We are working on the following pipeline: Neuronal network (AI) methods are being implemented, refined and trained for predicting low resolution structural features from a sequence of a protein of unknown structure. Currently we restrict the study to homologous membrane proteins. The predicted structural features will be converted to mechanical constraints (virtual potentials) and applied in a conformational search at reduced resolution. The structures of lowest virtual free energy will be selected and further minimised in all-atom molecular mechanics simulations. In these simulations experimental structural data will be also taken into account.
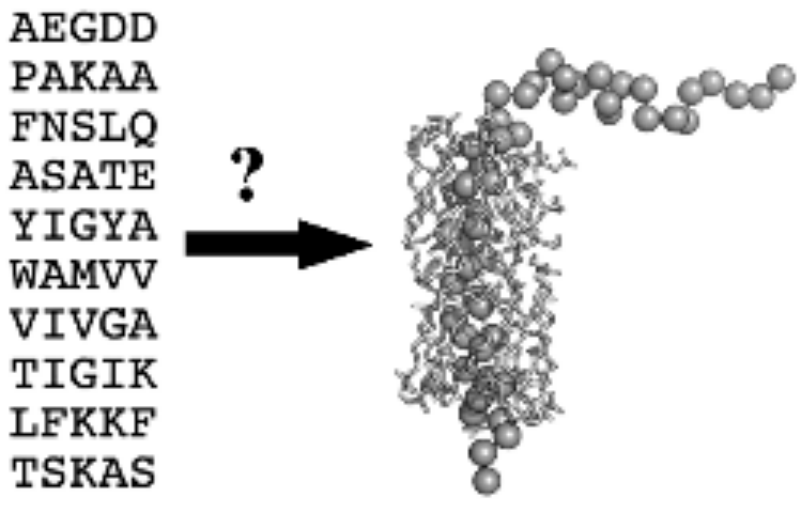
Figure 2. The sequence and the 3-dimensional fold of the major coat protein of the M13 bacteriophage, surrounded with the solvation or first shell lipids.
scientific adviser

research fellow

research fellow

research fellow

laboratory assistant
|
Tibor PÁLI
|
scientific adviser | publications | CV |
|
Krisztina SEBŐKNÉ NAGY
|
research fellow | publications | CV |
|
Teruaki KOTO
|
research fellow | publications | CV |
|
Zoltán KÓTA
|
research fellow | publications | CV |
|
Csilla BARTA
|
laboratory assistant |